
Las Malformaciones del Sistema Nerviosos Central (SNC) incluyen una amplia gama de patologías y síndromes las cuales tienen su origen en el periodo embrionario 16 a 28 días de gestación (cuando el embrión mide entre 1 a 5 milímetros. También el SNC es uno de los sistemas en el cual, se presentan alteraciones que pueden tener su origen en tiempo ulterior, como los problemas en algunas estructuras como el Cuerpo Calloso, así como la diferenciación y maduración del cerebro que finaliza en el periodo de la lactancia, por lo tanto es indispensable el cuidado que debe tener la mujer gestante en las primeras 8 semanas luego del coito fecundante, pues es el periodo crucial en la formación del Sistema Nervioso Central del bebé.
Debe asegurarse que en este periodo, los requerimientos de Ácido Fólico sean cubiertos (3 mgs diarios) , evitar procesos infecciosos ( rubéola , sarampión , citomegalovirus y otros) la presencia de tóxicos (venenos ) órganos fosforados de uso agrícola , alcohol , cocaína, y otros, cuyo efecto nocivo es variable de acuerdo a la respuesta individual existiendo personas con mayor predisposición a presentarlo que otros , esta tendencia personal no puede medirse por lo cual la prevención debe ser para todas las mujeres embarazadas.
Las malformaciones congénitas del SNC, pueden tener su origen en errores embriológicos en los diferentes pasos del desarrollo del Tubo Neural
1) Alteraciones de la Inducción Dorsal
(Anencefalia, Encefalocele-Iniencefalia, Meningocele, Mielomeningocele, Espina Bífida, Malformación de Arnold-Chiari , Regresión Caudal)
2) Alteraciones de la Inducción Ventral
Síndrome de Dandy Walker , Holoprosencefalia
3) Alteraciones de la Proliferación, y Diferenciación Neuronal
Microcefalia , Macrocefalia
4) Alteraciones de la Migración Neuronal
Agenesia del Cuerpo Calloso
5) Lesiones Adquiridas
Leucomalacia, Porencefalia, Ezquiscencefalia
MALFORMACIÓN DE ARNOLD-CHIAR

Descripción

 Este tipo, también denominado malformación de Chiari clásica o Malformación de Arnold-Chiari propiamente dicha, involucra la protrusión de estructuras cerebelosas y también del Tallo Cerebral a través del Foramen. A menudo el Vermis está, además, incompleto o ausente, y todo el cuadro suele acompañarse de Hidrocefalia y Mielomeningocele a nivel lumbar. Esta complicación puede derivar en parálisis parcial o total por debajo del Mielomeningocele.
Este tipo, también denominado malformación de Chiari clásica o Malformación de Arnold-Chiari propiamente dicha, involucra la protrusión de estructuras cerebelosas y también del Tallo Cerebral a través del Foramen. A menudo el Vermis está, además, incompleto o ausente, y todo el cuadro suele acompañarse de Hidrocefalia y Mielomeningocele a nivel lumbar. Esta complicación puede derivar en parálisis parcial o total por debajo del Mielomeningocele.
 Imagen radiológica de un paciente con malformación de Chiari tipo I, que muestra una hernia tonsilar de menos de 3 mm.
Imagen radiológica de un paciente con malformación de Chiari tipo I, que muestra una hernia tonsilar de menos de 3 mm.

 Tomografía Axial computada sagital de un recién nacido. En la imagen se observa la presencia de una malformación de Chiari del tipo I.
Tomografía Axial computada sagital de un recién nacido. En la imagen se observa la presencia de una malformación de Chiari del tipo I.
DISRAFIAS ESPINALES
( Encefalocele, Meningocele, Mielomeningocele, Espina Bífida)
Diastematomiela Espina bífida dorsal
Espina bífida dorsal
Meningocele (raquídeo)



 La tomografía axial computarizada (TAC) es un buen método para el estudio de la Disrafia Espinal, por los detalles anatómicos que permite identificar. Sin embargo, tiene como desventaja su mayor costo, el uso de radiación ionizante, la necesidad de sedación del paciente y en muchos casos la necesidad de inyectar medio de contraste intrarraquídeo.
La tomografía axial computarizada (TAC) es un buen método para el estudio de la Disrafia Espinal, por los detalles anatómicos que permite identificar. Sin embargo, tiene como desventaja su mayor costo, el uso de radiación ionizante, la necesidad de sedación del paciente y en muchos casos la necesidad de inyectar medio de contraste intrarraquídeo.
La US es el método de “screening” más indicado hoy en día, ante la sospecha de una Disrafia Espinal Oculta, para seleccionar los casos que requieran estudio más sofisticado. En especial, es el examen indicado en niños con manifestaciones cutáneas sugerentes de Disrafia Espinal.
ANOMALÍAS DE LA UNIÓN CRÁNEO-CERVICAL
Alteraciones óseas congénitas o adquiridas del hueso occipital, el agujero magno o las dos primeras vértebras cervicales que disminuyen el espacio virtual ocupado por el tronco del encéfalo y la médula espinal, dando lugar a síntomas cerebelosos, de los pares craneales inferiores y medulares.
Debido a la flexibilidad medular y su susceptibilidad a la compresión intermitente, las lesiones a este nivel pueden producir síntomas variables entre un paciente a otro y, pueden ser intermitentes.
La fusión del atlas y el hueso occipital produce síntomas de Mielopatía Cervical cuando el diámetro anteroposterior del agujero magno por detrás de la Apófisis Odontoides disminuye a 19mm.

La Platibasia es un aplanamiento asintomático de la base del cráneo, es decir, el ángulo formado por la intersección del plano del Clivus y el plano de la fosa anterior es 135° en la Rx lateral de cráneo. La impresión o invaginación basilar (protrusión de la apófisis odontoides dentro del agujero magno) produce el Síndrome de Cuello Corto y una combinación de signos cerebelosos, troncoencefálicos, de pares craneales bajos y medulares.

La malformación de Klippel-Feil (fusión de las vértebras cervicales) suele ser asintomática, excepto por la presencia de deformidad cervical con un grado de movilidad reducido.
La subluxación atloaxoidea (desplazamiento anterior del atlas en relación al Axis) produce compresión medular aguda o crónica.
Etiología
Anomalías congénitas. Incluyen el hueso Odontoideo, Asimilación e Hipoplasia del atlas y malformación de Chiari (descenso de amígdalas cerebelosas o el Vermis al interior del canal medular cervical.
Anomalías cerebrales
La Acondroplasia puede producir un estrechamiento del agujero magno y compresión de estructuras nerviosas. El Síndrome de Down, el Síndrome de Morquio (mucopolisacaridosis tipo IV) y la Osteogénesis imperfecta pueden causar una inestabilidad atloaxoidea y compresión medular secundaria.
Alteraciones adquiridas.
Pueden ser secundarias a traumatismos y a varias enfermedades.
La lesión traumática del Complejo Occipitoatloaxoideo tiene una elevada mortalidad en el lugar del accidente. Estas lesiones pueden ser óseas (fracturas), ligamentosas (luxaciones) o complejas (subluxación de C-2, lesión transaxial de la unión bulbo-medular y roturas osteoligamentosas).

La mitad de las mismas son causadas por accidentes de tráfico o bicicleta, el 25 por ciento por caídas y el 10 por ciento en actividades recreativas, particularmente accidentes por zambullida en albercas.
Una lesión cervical menor puede precipitar síntomas y signos progresivos variables en pacientes con una anomalía de la Unión Craneocervical subyacente. La AR y las lesiones metastásicas de la columna cervical pueden producir una subluxación atloaxoidea.
Un tumor de lento crecimiento (p. ej., meningioma, cordoma) en la unión craneocervical produce síntomas por invasión del tronco del encéfalo o la médula espinal. La AR y la Enfermedad de Paget pueden dar lugar a una impresión basilar con compresión del tronco o la médula. La AR es la causa más frecuente de inestabilidad craneocervical, la cual puede ser también secundaria a traumatismo, erosión tumoral o enfermedad de Paget.
Síntomas y signos
La presentación clínica varía debido a que las anomalías del hueso y los tejidos blandos pueden comprimir la médula cervical, el TE, los pares craneales, las raíces cervicales o su irrigación vascular en diferentes combinaciones. Es frecuente la existencia de una postura anómala de la cabeza y, en algunos pacientes, el cuello es corto o membranoso.
 Los síntomas más frecuentes son el dolor cervical y la compresión medular (Mielopatía). La compresión de las vías motoras produce debilidad muscular, espasticidad e hiperreflexia en los miembros superiores y/o inferiores. La afectación de la neurona motora inferior produce atrofia muscular y debilidad en brazos y manos. Las alteraciones sensitivas (incluidas la sensibilidad posicional articular y la vibratoria) suelen reflejar afectación de las columnas posteriores.
Los síntomas más frecuentes son el dolor cervical y la compresión medular (Mielopatía). La compresión de las vías motoras produce debilidad muscular, espasticidad e hiperreflexia en los miembros superiores y/o inferiores. La afectación de la neurona motora inferior produce atrofia muscular y debilidad en brazos y manos. Las alteraciones sensitivas (incluidas la sensibilidad posicional articular y la vibratoria) suelen reflejar afectación de las columnas posteriores.
Los pacientes pueden referir sensación de hormigueo con la flexión del cuello, que desciende por la espalda y a menudo por los brazos (signo de Lhermitte). La afectación de los haces espinotalámicos (p. ej., con pérdida de sensibilidad termoalgésica) es menos frecuente, pero puede manifestarse por parestesias o sensación de entumecimiento en «guante y en calcetín».
Los déficit del TE y los pares craneales pueden consistir en apnea del sueño, oftalmoplejia internuclear (debilidad de la aducción del ojo ipsilateral y nistagmo horizontal en la abducción del ojo contralateral en la mirada horizontal), nistagmo con componente rápido hacia abajo, ronquera, disartria y disfagia (debidas a debilidad e incoordinación de la lengua, paladar blando, faringe y laringe).
Es frecuente el dolor cervical que se extiende a los brazos y la cefalea suboccipital irradiada al vértice del cráneo. Los síntomas empeoran con los movimientos de la cabeza y pueden precipitarse con la tos o al inclinarse hacia delante. El dolor se atribuye a compresión de la raíz C-2 y el nervio occipital mayor y a disfunción musculoesquelética local.
Los síntomas vasculares incluyen síncope, drop attacks, vértigo, períodos intermitentes de confusión y alteración del nivel de conciencia, debilidad muscular episódica y alteraciones visuales transitorias. El movimiento o cambio de posición de la cabeza puede precipitar una isquemia vertebrobasilar.
Diagnóstico
Debe considerarse una Anomalía Craneocervical cuando existen déficit neurológicos fijos o progresivos del tronco del encéfalo, la médula cervical alta o el cerebelo.
La Rx (lateral de cráneo abarcando la columna cervical y anteroposterior y oblicua de la columna) se utilizan para identificar factores que pueden influir en el tratamiento. Estos factores incluyen la reductibilidad de una lesión (capacidad de conseguir un alineamiento óseo normal y eliminar con ello la compresión de estructuras nerviosas), erosión ósea, mecánica de la compresión y presencia de centros de osificación anormales o placas de crecimiento epifisario con desarrollo anómalo.
La TC con contraste intratecal proporciona detalles anatómicos de la alteración de la estructura nerviosa y la distorsión ósea asociada. La RM sagital identifica mejor las lesiones nerviosas (herniación cerebral caudal, siringomielia y alteraciones vasculares). La RM permite correlacionar la patología ósea o de tejidos blandos, definir el nivel y la extensión de una malformación y el defecto neural asociado (p. ej., malformación de Chiari, siringomielia). La Angiografía Vertebral o la angio-resonancia se utilizan selectivamente para identificar un compromiso vascular fijo o dinámico.

Tratamiento
Algunas alteraciones de la Unión Craneocervical (p. ej., luxación atloaxoidea aguda traumática y lesiones ligamentosas agudas) pueden reducirse y realinearse únicamente con la colocación de la cabeza, aliviando la compresión de estructuras nerviosas. La mayor parte de los casos requieren tracción esquelética con un aro en corona o halo con incrementos graduales de hasta 3 a 3,5 kg para conseguir la reducción.
La tracción suele ser eficaz en 5 a 6 días. Si se consigue la reducción, se mantiene un chaleco con halo durante 8 a 12 semanas; posteriormente se realiza una nueva Rx para confirmar la estabilidad. Si la reducción no elimina la compresión está indicada la descompresión quirúrgica mediante abordaje anterior o posterior. Si la inestabilidad continúa después de la descompresión, es necesaria una fijación posterior. En otros casos (como en la AR) la inmovilización externa aislada no suele conseguir la reducción permanente, requiriendo habitualmente una fijación posterior (estabilización) o descompresión anterior y estabilización.
Existen varias técnicas de fusión en la región craneocervical. En general, se fusionan todos los niveles inestables. La instrumentación proporciona una estabilidad inmediata, hasta que se desarrolla la fusión ósea, y duradera.
En las metástasis puede ser útil la radioterapia y la inmovilización con collarín cervical. En los pacientes con Enfermedad de Paget pueden ayudar la calcitonina, mitramicina y bifosfonatos.
MÉDULA ANCLADA Y SIRINGOMIELIA

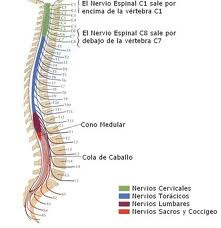 Al nacimiento, el Cono Medular se encuentra aproximadamente al nivel de los cuerpos vertebrales T12 a L3. Hay un importante ascenso de la médula espinal durante los primeros 2 meses de vida.
Al nacimiento, el Cono Medular se encuentra aproximadamente al nivel de los cuerpos vertebrales T12 a L3. Hay un importante ascenso de la médula espinal durante los primeros 2 meses de vida.
 Las radiografías simples muestran con frecuencia espina bífida en L4, L5 ó S1 pero su ausencia no descarta el diagnóstico. La resonancia magnética de la Columna Lumbosacra es el examen de elección.
Las radiografías simples muestran con frecuencia espina bífida en L4, L5 ó S1 pero su ausencia no descarta el diagnóstico. La resonancia magnética de la Columna Lumbosacra es el examen de elección.
En la Siringomielia Postraumática, se forma un quiste o cavidad llena de líquido dentro de la médula. Esta actividad se puede expandir con el transcurso del tiempo, extendiéndose dos o más segmentos espinales a partir del nivel de la lesión medular.
La Siringomielia también se produce en personas con una Anomalía Cerebral congénita llamada Malformación de Chiari: durante el desarrollo del feto, la parte inferior del cerebelo sobresale de la parte posterior de la cabeza hacia la porción cervical del canal espinal. Los síntomas habitualmente incluyen vómitos, debilidad muscular en la cara y la cabeza, dificultad para tragar y diversos grados de alteración mental.
 La Siringomielia también puede estar asociada con la Espina Bífida, tumores en la médula espinal, aracnoiditis y siringomielia idiopática (de causa desconocida). Las IRM han aumentado significativamente la cantidad de diagnósticos en las primeras etapas de Siringomielia. Los signos del trastorno tienden a desarrollarse con lentitud, aunque se puede producir un inicio repentino con la tos o el esfuerzo excesivo.
La Siringomielia también puede estar asociada con la Espina Bífida, tumores en la médula espinal, aracnoiditis y siringomielia idiopática (de causa desconocida). Las IRM han aumentado significativamente la cantidad de diagnósticos en las primeras etapas de Siringomielia. Los signos del trastorno tienden a desarrollarse con lentitud, aunque se puede producir un inicio repentino con la tos o el esfuerzo excesivo.
La cirugía produce la estabilización o una modesta mejora de los síntomas para la mayoría de las personas. La demora en realizar un tratamiento puede resultar en una lesión irreversible de la médula espinal. La recurrencia de la Siringomielia después de la cirugía puede hacer necesarias otras operaciones; éstas pueden no ser completamente exitosas a largo plazo. Hasta la mitad de las personas tratadas por Siringomielia tienen retorno de síntomas dentro de los cinco años.
MALFORMACIÓN DE ARNOLD-CHIAR
La Malformación de Chiari, malformación de Arnold-Chiari o Anormalidad de Chiari, consiste en un defecto estructural o ubicación anormal del cerebelo en la base del cráneo, con descenso del Vermis por el Foramen Magnum, que puede verse acompañado de Hidrocefalia y Mielomeningocele Lumbar.
La malformación fue descripta por primera vez en 1883 por el doctor Cleland, en un artículo titulado “Contribution to the study of spina bifida, encephalocele, and anencephalus” (-Contribución al estudio de la espina bífida, el encefalocele y la anencefalia-) en el Journal of Anatomy and Physiology. Ocho años más tarde, el Neurólogo austríaco Hans Chiari escribió un artículo en el Deutsche Medizinische Wochenschriff titulado "Acerca de las alteraciones cerebelosas resultantes de la hidrocefalia cerebral", en el cual reconocía el trabajo de Cleland y añadía su propio sistema de clasificación de este desorden en tipos I, II y III.
En 1896, Chiari definió la enfermedad de la siguiente forma: "Una elongación en forma de cuña de las amígdalas cerebelosas y de la parte medial de los lóbulos inferiores del cerebelo, que corren a lo largo de la médula dentro del canal cervical".
En 1907, Schwalbe y Gredig, dos estudiantes de un equipo de la Universidad de Heidelberg liderado por el doctor Julius Arnold, describieron un total de otros cuatro casos, anteponiendo el nombre de su maestro al de Chiari en la designación de la malformación .
Descripción
Hans Chiari
Las malformaciones de Chiari consisten en defectos estructurales del cerebelo, órgano encargado de controlar el equilibrio y la coordinación neuromuscular. El cerebelo normal ocupa un espacio indentado en la parte inferior del hueso occipital, encima del Foramen Magnum, orificio por el cual desciende el extremo del tronco encefálico hacia el Canal Medular. La malformación de Chiari se produce cuando parte del cerebelo se encuentra ubicado por debajo del Foramen.
Si la apertura del espacio óseo que debería ocupar el cerebelo es menor que lo normal, el Cerebelo y el Tallo Cerebral pueden verse empujados hacia abajo y obligados a avanzar sobre la sección superior del canal espinal. La presión resultante sobre estas estructuras nerviosas puede afectar las funciones que llevan a cabo, y entorpecer o bloquear total o parcialmente el flujo del Líquido Cefalorraquídeo (LCR) hacia el cerebro.
Etiología
No se conocen las causas exactas de las malformaciones de Chiari. Sin embargo, se han sugerido diferentes mecanismos de formación, los que llevan al primero y más básico sistema de clasificación: Chiari primarias y Chiari secundarias.
Malformaciones de Chiari primarias
Una o varias fallas en el desarrollo embrionario pueden conducir a las Malformaciones de Chiari. Las sospechas más probables, son ciertas mutaciones genéticas o la falta de nutrientes o vitaminas en la dieta materna.
El hecho de que unos pocos pacientes de esta patología tengan antecedentes familiares de la misma malformación, sugiere que algunos casos pueden ser hereditarios.
Malformaciones de Chiari secundarias
Se producen cuando, en una etapa posterior de la vida, una infección, traumatismo o intoxicación produce un drenaje excesivo de LCR desde las regiones lumbares o torácicas hacia el Encéfalo y empujando al Cerebelo hacia abajo, resultando entonces en una Chiari adquirida o secundaria. Estas últimas son, con mucha diferencia, más raras que las anteriores.
Clasificación
Diagrama de una malformación de Chiari tipo II donde se indican los distintos puntos potenciales de obstrucción que llevan a diferentes clases de hidrocefalia….pie de foto.
Estas patologías se clasifican clínicamente por la severidad de los síntomas y anatómicamente por las partes del encéfalo que se prolongan en la cavidad espinal. Los tres primeros tipos fueron descriptos por el propio Hans Chiari.
Tipo I
Si las amígdalas Cerebelosas son empujadas al canal medular sin involucrar al tallo cerebral, se considera que la Chiari es del Tipo I. Afortunadamente, no suele causar síntomas, ya que es el tipo más frecuente. Normalmente solo se diagnostica en la adultez, durante un examen dirigido a diagnosticar otras patologías. Todas las Chiari adquiridas o secundarias pertenecen exclusivamente al Tipo I.
Las malformaciones de Chiari tipo I pueden, no obstante, obstruir el flujo de líquido cefalorraquídeo y comprimir la médula espinal. Algunas veces están asociadas a la Siringomielia.
Tipo II

Tipo III
La forma más grave de la anormalidad. El Cerebelo y el Tallo Cerebral, herniados, se adentran en el Canal Medular Cervical, a menudo acompañados del Cuarto Ventrículo Cerebral y comprimen la Médula Espinal. En algunos casos raros, el tejido cerebeloso herniado asoma por una abertura anormal del hueso occipital, formando una bolsa llamada Encefalocele Occipital, que puede contener incluso tejido cerebral. Este tipo de Chiari produce serios síntomas neurológicos.
Tipo IV
El tipo IV, de descripción más reciente, involucra un desarrollo incompleto de las estructuras del cerebelo, enfermedad conocida como Hipoplasia Cerebelosa. Esta forma es rara, y puede involucrar amígdalas muy bajas en el canal espinal, estructuras cerebelosas ausentes, y partes del cráneo y la médula espinal al descubierto.
[editar]
Tipo 0
Esta nueva forma de la enfermedad, aún se encuentra en discusión, no existe protrusión del cerebelo a través del Foramen Magno, pero a pesar de ello, los síntomas neurológicos se encuentran presentes.
Prevalencia
Durante el siglo XIX y principios del XX, la frecuencia o estadística de la enfermedad se estimaba en 1:1000 (uno en mil nacidos vivos). Sin embargo, el uso de métodos diagnósticos por imágenes ha demostrado que las Malformaciones de Chiari son mucho más comunes.
El diagnóstico se dificulta aún más por el hecho de que la mayoría de los infantes nacidos con esta enfermedad no manifiestan síntomas hasta la adolescencia, la adultez o la vejez, si es que alguna vez evidencian alguno.
Las Chiari son más frecuentes en las mujeres que en los hombres, y el tipo II es más común en ciertos grupos étnicos, como los de ascendencia céltica.
Síntomas
La naturaleza variable de la malformación conduce a un amplísimo abanico de signos y síntomas, que muchas veces complican o confunden el diagnóstico.
Sin embargo, el síntoma más común es el dolor de cabeza, normalmente localizado en el área occipital y extendido hacia la calota craneana, agravándose al toser, agacharse o hacer esfuerzos físicos. De hecho, el dolor en la parte posterior de la cabeza es el síntoma primario —y muchas veces el único— de la mayoría de los pacientes.
La enorme colección de signos y síntomas posibles en la Enfermedad de Chiari se agrupa de la siguiente manera:
Craneales
El mencionado dolor de cabeza occipital.
Problemas relacionados con los pares craneales: disfagia, hipo, ronquera por mucosidad laríngea, síncope, asfixia por aspiración mucosa, dolor o insensibilidad facial, ausencia del reflejo del vómito, pérdida sensorial del nervio trigémino par, parálisis de las cuerdas vocales y apnea.
Espinales
*Fallas motrices: incontinencia, espasticidad atrófica, incontinencia, debilidad muscular, hemiparesia, cuadriparesia, hipotonía, atrofia.
*Fallas sensitivas: temblores, anestesia, dolor, sensación de quemadura, dolor o anestesia no radicular del tronco, los hombros y los miembros, pérdida sensitiva disociada, parestesia, hiperestesia, disestesia, falta de sentido de la posición espacial, problemas de temperatura, articulaciones de Charcot.
*Otros problemas medulares: pérdida del equilibrio, torpeza, hiporreflexia, arreflexia, ataxia, hiperreflexia, dismetría, clono y signo de Babinski.
Oculares
Dolor o presión ocular, fotofobia, problemas de campo visual, falta de agudeza, paresias de los músculos extraoculares y nistagmo.
Otorrinolaringológicos
Hipoacusia, mareos, desequilibrio, vértigos, ruido de fondo, sensación de presión en el oído.
Otros
Escoliosis, síndrome de Klippel-Feil, Hidrocefalia, fatiga, dolor esquelético axial, anomalía osciloscópica asociada a problemas de la base del cráneo.
Diagnóstico
La enorme mayoría de los pacientes de Malformación de Chiari no presentan síntomas o, cuando los presentan, son de una naturaleza sumamente inespecífica, como se observa en el listado anterior. Por consiguiente, la malformación sólo suele ser correctamente diagnosticada durante procedimientos diagnósticos o terapéuticos destinados a otras enfermedades.
Cuando se sospecha la presencia de una Chiari, puede procederse a los siguientes métodos diagnósticos:
* Interrogatorio clínico y examen físico orientados a verificar la memoria del paciente, su capacidad cognoscitiva, el equilibrio, reacciones, reflejos, sensibilidad y habilidad motriz.
* Resonancia magnética, también llamada RM o IRM. Manifiesta al 100% tipo y grado de Chiari.
* Rayos X de cabeza y cuello.
* Tomografía axial computada o TAC.
Uno o varios de estos métodos —bien utilizados— son capaces de diagnosticar adecuadamente la presencia de una Chiari y de identificar el tipo al que pertenece.
Tratamiento

Muchos pacientes de Chiari son asintomáticos y, como tales, no requieren tratamiento. En otros casos, el tratamiento es sintomático —típicamente, analgésicos para aliviar el dolor—.
Si se observan perturbaciones funcionales o progreso de la enfermedad, el único tratamiento posible es la cirugía correctiva. La mayoría de los pacientes observan grandes mejorías o reducción de la gravedad de sus síntomas luego de la operación, pudiendo ser necesarias más de una para aliviar el cuadro.
En los adultos, puede practicarse Cirugía Descompresiva de la Fosa Posterior para generar más espacio para el cerebelo y aliviar la presión sobre la médula y la columna superior. Se lleva a cabo abriendo un orificio en la parte posterior de la base del cráneo y retirando una porción de esta —y a veces de la columna—, para corregir la estructura ósea anormal. Mediante un Electrocauterio se eliminan las porciones anómalas del cerebelo.
Un procedimiento similar, denominado Laminectomía Espinal, extirpa la lámina (parte del canal espinal) para aumentar el diámetro de este y descomprimir la médula y las raíces nerviosas, creando de paso más espacio para normalizar el flujo del LCR.
Los bebés o niños con Mielomeningocele suelen requerir cirugía para cerrar la abertura posterior y volver a colocar el tejido medular en su sitio. Los Hidrocéfalos exigen el implante de un sistema de válvulas que drenan el exceso de LCR y restablecen la presión intracraneal normal. Se coloca un tubo en la cabeza que lleva el exceso de fluido hacia la pared torácica o abdominal, donde puede ser reabsorbido.
Una técnica alternativa es la Tercerventriculotomía, mediante la cual se practica un pequeño orificio en el piso del tercer ventrículo para llevar el exceso de flujo hacia el espacio subaracnoideo.
Si el cuadro incluye Siringomielia o Hidromielia, se abrirá la médula espinal y —como en los casos anteriores— se insertará un catéter en la siringe para desviar el flujo de LCR.
Pronóstico
Si los procedimientos quirúrgicos indicados son realizados por un neurocirujano competente y con experiencia en Malformaciones de Chiari, el pronóstico de la enfermedad es excelente. El estado del paciente a largo plazo luego de la operación es de mejor a bueno en un 92 por ciento de los niños y en el 88 por ciento de los adultos. Solo el 8 por cineto de los pacientes pediátricos y el 12 por ciento de los adultos no evidencian mejoría tras una cirugía exitosa.
“Cirugía exitosa" se define, en estos casos, como aquella que ha conseguido restablecer y normalizar el flujo de LCR. Las causas de la falta de mejoría en algunos pacientes tras una cirugía exitosa pueden ser varias, como por ejemplo la formación de excesivo tejido cicatrizal. En el resto de los casos, el restablecimiento es completo y casi siempre se observa la desaparición de todos los síntomas.
DISRAFIAS ESPINALES
( Encefalocele, Meningocele, Mielomeningocele, Espina Bífida)
Las Disrafias Espinales son un grupo de patologías que se caracterizan por una anomalía en el desarrollo del Tubo Neural. Se clasifican en dos grupos: Disrafias Abiertas y Disrafias Ocultas o Cerradas.
Las primeras corresponden a malformaciones precoces en el desarrollo embrionario de las estructuras medulares y raquídeas y, en todas ellas, las estructuras nerviosas y Meníngeas se encuentran comunicadas con el medio externo, lo que hace que su corrección quirúrgica sea urgente.
Patologías incorporadas
Quedan incluidas las siguientes enfermedades y los sinónimos que las designen en la terminología médica habitual:
Diastematomiela
Encefalocele de cualquier sitio
Encefalocele frontal
Encefalocele nasofrontal
Encefalocele occipital
Encefalocele
Encefalomielocele
Espina bífida (abierta) (quística)
Espina bífida cervical con hidrocefalia
Espina bífida cervical sin hidrocefalia
Espina bífida con hidrocefalia
Espina bífida dorsal con hidrocefalia
Espina bífida lumbar con hidrocefalia
Espina bífida lumbar sin hidrocefalia
Espina bífida lumbosacra
Espina bífida lumbosacra con hidrocefalia
Espina bífida oculta
Espina bífida sacra con hidrocefalia
Espina bífida sacra sin hidrocefalia
Espina bífida torácica con hidrocefalia
Espina bífida torácica sin hidrocefalia
Espina bífida toracolumbar
Espina bífida toracolumbar con hidrocefalia
Espina bífida, no especificada
Filum Corto
Filum Corto
Hidroencefalocele
Hidromeningocele (raquídeo)
Hidromeningocele craneano
Lipoma Cono Medular
Lipoma D Efilum
Lipomeningocele
Lipoma Extradural
Médula Anclada
Médula Anclada
Mielocele
Mielomeningocele
Mielocistocele
Mielomeningocistocele
Meningocele (raquídeo)
Meningocele cerebral
Meningoencefalocele
Meningomielocele
Quiste Dermoide o Epidermode Raquideo
Quiste Neuroentrico
Raquisquisis
Siringomielocele
Sinus Dermal
Hay múltiples malformaciones de la médula espinal, que se describen como parte de las Disrafias Espinales (meningocele, mielomeningocele, mielosquisis, espina bífida oculta, diastematomelia, lipoma intraespinal, lipomielomeningocele, filum terminal corto, bandas fibrosas, quiste dermoide intratecal), la mayoría de ellas asociadas a médula anclada. Este tipo de lesiones son más frecuentes en mujeres y pueden tener asociación familiar.

Para el Neurólogo Pediatra lo más importante es detectar la lesión, para así prevenir el daño neurológico progresivo, irreversible que produce la médula anclada. Este anclamiento tracciona la médula, posicionándola en un nivel anormalmente bajo.
En el feto, la médula espinal y el canal espinal tienen la misma longitud, el crecimiento diferente que experimentan estas estructuras en forma normal lleva a un ascenso relativo del cono medular. Al nacer, este está ubicado en L2-L3, alcanzando el nivel adulto del cono (L1-L2) a los 2 o 3 meses de edad. Al flexionar la columna normal, se produce un ascenso del cono; si el cono está anclado sufre daño por tracción repetida, lo que lleva a daño progresivo del segmento inferior de la médula espinal. El Síndrome de la Médula Anclada se presenta clínicamente en promedio a los 3 años de edad y se manifiesta en la mayoría de los casos con alteraciones sensitivas y motoras de las extremidades inferiores, que conducen a deformaciones ortopédicas.
Son comunes otros síntomas y signos, tales como lumbago, acortamiento de extremidades inferiores, cojera, escoliosis y cambios tróficos de la piel. Un 20 por ciento de los pacientes presentan vejiga neurogénica, que se manifiesta como enuresis o infecciones urinarias repetidas. Una vez que aparecen estos síntomas, la mayoría de estos son irreversibles. Por este motivo, el diagnóstico de estas condiciones debe ser precoz, es decir, antes que se desarrollen síntomas neurológicos.
Los marcadores cutáneos pueden ser la única alteración, especialmente en el recién nacido, que nos indique que puede existir una Disrafia Espinal Oculta. Su diagnóstico oportuno y el tratamiento quirúrgico precoz, consistente en liberar la médula espinal, previenen el daño neurológico o evitan su progresión.
Las lesiones cutáneas congénitas de la línea media deben alertar al pediatra, dado que estas pueden ser marcadoras de una malformación de la médula espinal. La mayoría de las lesiones se encuentra alrededor de la línea media y en la región lumbo-sacra, aunque lesiones en la región torácica o cervical también han sido descritas como indicadores de una malformación subyacente.
En la literatura se describen lesiones cutáneas en la región lumbo-sacra del 48 al 100 por ciento de los pacientes que presentan Disrafia Espinal. Por el contrario, no hay datos confiables de qué porcentaje de lesiones cutáneas realmente son indicadoras de patología espinal.
Hay ciertas lesiones que se describen como de alta sospecha, como Hipertricosis, Hoyuelo o Seno Dérmico, Papiloma o Pseudocola, Lipoma, Hemangioma, Aplasia Cutis, Quiste Dermoides y otras que se describen como de baja sospecha, que incluyen Telangiectasia, Malformación Capilar, Hiperpigmentación, Nevo Melanocítico y Teratoma.
Dentro de estas, la Hipertricosis puede ser localizada, discreta o como un acúmulo mayor de pelos, como ocurre en el Nevo en Cola de Fauno, que se presenta con pelo claro u oscuro y textura generalmente sedosa.
Cuando existe una gran cantidad de pelos se asocia generalmente a otros estigmas cutáneos y es altamente indicativo de Disrafia Espinal. También puede existir pelo escaso como un hallazgo normal, lo cual hace difícil la decisión entre realizar o no estudio completo.
Los Lipomas, cuando son congénitos, son altamente sugerentes de lesión espinal; pueden estar ubicados en la dermis, en el canal espinal o ser parte del saco que se hace camino a través del defecto vertebral (lipomielomeningocele) . El estudio radiológico debe ser exhaustivo, dado que hay conexiones intraespinales mínimas que deben ser descartadas o diagnosticadas.
Los Hemangiomas se asocian a Disrafia Espinal en general cuando son grandes (mayores de 4 cm), muchos sobrepasan la línea media y presentan una erosión o ulceración en su superficie. Cuando son pequeños, la asociación con Disrafia Espinal es menos frecuente.
Las Telangiectasias y Malformaciones capilares, cuando son el único hallazgo, raramente se asocian a Disrafia Espinal, pero la relación costo-beneficio indicaría de todas maneras hacer estudio radiológico.
Los Orificios Cutáneos, como los senos dérmicos, se localizan con mayor frecuencia en la región lumbo-sacra o en la región cervical y son de los marcadores congénitos de Disrafia Espinal mejor establecidos. Son orificios profundos que en la región lumbosacra se ubican más arriba del pliegue interglúteo y en su mayoría se comunican con el canal raquídeo y pueden ser causa de meningitis a repetición. Por el contrario, aquellos orificios localizados en la región sacro-coccígea y cubiertos por el pliegue interglúteo corresponden a senos o fosetas pilonidales, que en su gran mayoría no se comunican con el canal raquídeo y no tienen significado patológico.
La Aplasia Cutis se define como la ausencia congénita de piel y es más frecuente en el cuero cabelludo. Algunas formas de Aplasia Cutis (específicamente la forma membranosa) serían secundarias a un cierre incompleto de las líneas de fusión embrionarias.
La localización lumbo-sacra se ha descrito con baja frecuencia, pero a menudo asociada a Disrafia Espinal. Se ha sugerido realizar estudio en Aplasia Cutis ubicadas posterior al vértex o en la línea media y no realizarlo en lesiones ubicadas anterior al vértex y laterales a la línea media. Las cicatrices congénitas de ubicación lumbo-sacra también se han descrito como marcadores de Disrafia y podrían ser una variante evolutiva de Aplasia Cutis.
Los papilomas, colas y pseudocolas constituyen tres lesiones que se asocian a Disrafia Espinal, por lo que se debe hacer estudio radiológico preoperatorio. El papiloma o acrocordón es un tallo compuesto por dermis, cubierto por epidermis. Una cola humana verdadera (vestigio persistente), en cambio, presenta un núcleo central con tejido graso maduro, vasos sanguíneos pequeños, haces de fibras musculares y fibras nerviosas. Una pseudocola es considerada un hamartoma compuesto por tejido graso y ocasionalmente por cartílago. Clínicamente estas lesiones son indistinguibles entre sí.
El paciente con un marcador cutáneo sospechoso debe ser evaluado en su contexto general, especialmente en un niño mayor. Se deben evaluar otras malformaciones, historia familiar de defectos del Tubo Neural, debilidad y/o dolor de extremidades inferiores, marcha alterada, escoliosis, incontinencia o dificultad para aprender a controlar esfínteres, infección urinaria recurrente o meningitis a repetición. Los glúteos deben ser simétricos y el pliegue interglúteo debe ser examinado especialmente en busca de orificios o papilomas. Si este pliegue se curva en su final superior, sugiere una masa subyacente, como podría ser un lipoma. Las extremidades inferiores deben ser examinadas en busca de cambios tróficos secundarios a daño neural.
Se dispone actualmente de varios métodos de diagnóstico por imágenes para estudiar una Disrafia Espinal. La radiografía simple de columna vertebral tiene bajo rendimiento, especialmente en las formas ocultas y en menores de un año, debido a la escasa osificación de los elementos vertebrales posteriores a esta edad. Puede no mostrar alteraciones aun cuando existan defectos en los elementos posteriores de la columna.
La resonancia magnética (RM) es una técnica que no usa radiación y permite una excelente representación de las alteraciones anatómicas en los casos de Disrafia Espinal, en especial porque permite efectuar cortes en cualquier orientación. Sus desventajas son el alto costo, la necesidad de sedar a los niños menores y su falta de disponibilidad en muchos centros de nuestro país. Este es en la actualidad el examen de mayor rendimiento en el diagnóstico de disrafia espinal.
La Ultrasonografía (US) es una técnica no invasiva, rápida, inocua, no necesita sedación ni medio de contraste, está disponible en la mayoría de los centros, es de bajo costo, se puede repetir las veces que se estime conveniente y permite excelente visualización de las estructuras intra o extrarraquídeas en niños menores de 18 o 24 meses. Su alto rendimiento se basa en la osificación incompleta de los elementos posteriores de la columna vertebral, que normalmente presentan los niños de esta edad, especialmente en el recién nacido y en el lactante menor. Su principal desventaja con respecto a la TAC y la RM es su menor resolución anatómica y espacial que puede ser de importancia en aquellos casos que requieran cirugía.
ANOMALÍAS DE LA UNIÓN CRÁNEO-CERVICAL
Alteraciones óseas congénitas o adquiridas del hueso occipital, el agujero magno o las dos primeras vértebras cervicales que disminuyen el espacio virtual ocupado por el tronco del encéfalo y la médula espinal, dando lugar a síntomas cerebelosos, de los pares craneales inferiores y medulares.
Debido a la flexibilidad medular y su susceptibilidad a la compresión intermitente, las lesiones a este nivel pueden producir síntomas variables entre un paciente a otro y, pueden ser intermitentes.
La fusión del atlas y el hueso occipital produce síntomas de Mielopatía Cervical cuando el diámetro anteroposterior del agujero magno por detrás de la Apófisis Odontoides disminuye a 19mm.
La Platibasia es un aplanamiento asintomático de la base del cráneo, es decir, el ángulo formado por la intersección del plano del Clivus y el plano de la fosa anterior es 135° en la Rx lateral de cráneo. La impresión o invaginación basilar (protrusión de la apófisis odontoides dentro del agujero magno) produce el Síndrome de Cuello Corto y una combinación de signos cerebelosos, troncoencefálicos, de pares craneales bajos y medulares.
La malformación de Klippel-Feil (fusión de las vértebras cervicales) suele ser asintomática, excepto por la presencia de deformidad cervical con un grado de movilidad reducido.
La subluxación atloaxoidea (desplazamiento anterior del atlas en relación al Axis) produce compresión medular aguda o crónica.
Etiología
Anomalías congénitas. Incluyen el hueso Odontoideo, Asimilación e Hipoplasia del atlas y malformación de Chiari (descenso de amígdalas cerebelosas o el Vermis al interior del canal medular cervical.
Anomalías cerebrales
La Acondroplasia puede producir un estrechamiento del agujero magno y compresión de estructuras nerviosas. El Síndrome de Down, el Síndrome de Morquio (mucopolisacaridosis tipo IV) y la Osteogénesis imperfecta pueden causar una inestabilidad atloaxoidea y compresión medular secundaria.
Alteraciones adquiridas.
Pueden ser secundarias a traumatismos y a varias enfermedades.
La lesión traumática del Complejo Occipitoatloaxoideo tiene una elevada mortalidad en el lugar del accidente. Estas lesiones pueden ser óseas (fracturas), ligamentosas (luxaciones) o complejas (subluxación de C-2, lesión transaxial de la unión bulbo-medular y roturas osteoligamentosas).
La mitad de las mismas son causadas por accidentes de tráfico o bicicleta, el 25 por ciento por caídas y el 10 por ciento en actividades recreativas, particularmente accidentes por zambullida en albercas.
Una lesión cervical menor puede precipitar síntomas y signos progresivos variables en pacientes con una anomalía de la Unión Craneocervical subyacente. La AR y las lesiones metastásicas de la columna cervical pueden producir una subluxación atloaxoidea.
Un tumor de lento crecimiento (p. ej., meningioma, cordoma) en la unión craneocervical produce síntomas por invasión del tronco del encéfalo o la médula espinal. La AR y la Enfermedad de Paget pueden dar lugar a una impresión basilar con compresión del tronco o la médula. La AR es la causa más frecuente de inestabilidad craneocervical, la cual puede ser también secundaria a traumatismo, erosión tumoral o enfermedad de Paget.
Síntomas y signos
La presentación clínica varía debido a que las anomalías del hueso y los tejidos blandos pueden comprimir la médula cervical, el TE, los pares craneales, las raíces cervicales o su irrigación vascular en diferentes combinaciones. Es frecuente la existencia de una postura anómala de la cabeza y, en algunos pacientes, el cuello es corto o membranoso.
Los pacientes pueden referir sensación de hormigueo con la flexión del cuello, que desciende por la espalda y a menudo por los brazos (signo de Lhermitte). La afectación de los haces espinotalámicos (p. ej., con pérdida de sensibilidad termoalgésica) es menos frecuente, pero puede manifestarse por parestesias o sensación de entumecimiento en «guante y en calcetín».
Los déficit del TE y los pares craneales pueden consistir en apnea del sueño, oftalmoplejia internuclear (debilidad de la aducción del ojo ipsilateral y nistagmo horizontal en la abducción del ojo contralateral en la mirada horizontal), nistagmo con componente rápido hacia abajo, ronquera, disartria y disfagia (debidas a debilidad e incoordinación de la lengua, paladar blando, faringe y laringe).
Es frecuente el dolor cervical que se extiende a los brazos y la cefalea suboccipital irradiada al vértice del cráneo. Los síntomas empeoran con los movimientos de la cabeza y pueden precipitarse con la tos o al inclinarse hacia delante. El dolor se atribuye a compresión de la raíz C-2 y el nervio occipital mayor y a disfunción musculoesquelética local.
Los síntomas vasculares incluyen síncope, drop attacks, vértigo, períodos intermitentes de confusión y alteración del nivel de conciencia, debilidad muscular episódica y alteraciones visuales transitorias. El movimiento o cambio de posición de la cabeza puede precipitar una isquemia vertebrobasilar.
Diagnóstico
Debe considerarse una Anomalía Craneocervical cuando existen déficit neurológicos fijos o progresivos del tronco del encéfalo, la médula cervical alta o el cerebelo.
La Rx (lateral de cráneo abarcando la columna cervical y anteroposterior y oblicua de la columna) se utilizan para identificar factores que pueden influir en el tratamiento. Estos factores incluyen la reductibilidad de una lesión (capacidad de conseguir un alineamiento óseo normal y eliminar con ello la compresión de estructuras nerviosas), erosión ósea, mecánica de la compresión y presencia de centros de osificación anormales o placas de crecimiento epifisario con desarrollo anómalo.
Tratamiento
Algunas alteraciones de la Unión Craneocervical (p. ej., luxación atloaxoidea aguda traumática y lesiones ligamentosas agudas) pueden reducirse y realinearse únicamente con la colocación de la cabeza, aliviando la compresión de estructuras nerviosas. La mayor parte de los casos requieren tracción esquelética con un aro en corona o halo con incrementos graduales de hasta 3 a 3,5 kg para conseguir la reducción.
La tracción suele ser eficaz en 5 a 6 días. Si se consigue la reducción, se mantiene un chaleco con halo durante 8 a 12 semanas; posteriormente se realiza una nueva Rx para confirmar la estabilidad. Si la reducción no elimina la compresión está indicada la descompresión quirúrgica mediante abordaje anterior o posterior. Si la inestabilidad continúa después de la descompresión, es necesaria una fijación posterior. En otros casos (como en la AR) la inmovilización externa aislada no suele conseguir la reducción permanente, requiriendo habitualmente una fijación posterior (estabilización) o descompresión anterior y estabilización.
Existen varias técnicas de fusión en la región craneocervical. En general, se fusionan todos los niveles inestables. La instrumentación proporciona una estabilidad inmediata, hasta que se desarrolla la fusión ósea, y duradera.
En las metástasis puede ser útil la radioterapia y la inmovilización con collarín cervical. En los pacientes con Enfermedad de Paget pueden ayudar la calcitonina, mitramicina y bifosfonatos.
MÉDULA ANCLADA Y SIRINGOMIELIA
El Síndrome de Médula Anclada (SMA) es una malformación congénita en la que se produce una fijación de la médula dentro del canal espinal. Clínicamente se caracteriza por afectación musculoesquelética, cutánea, neurológica y urológica. El diagnóstico se basa en la clínica y en la resonancia magnética (RM) lumbar. Suele diagnosticarse en la infancia, aunque algunos pacientes inician los síntomas en la edad adulta.

El Síndrome de Médula Anclada es una enfermedad poco frecuente y de diagnóstico muy tardío en el adulto. Por su sintomatología insidiosa e inespecífica hay que considerarlo en el diagnóstico diferencial de los síndromes medulares y de las polineuropatías dada su potencial reparación quirúrgica.
El Síndrome de Médula Espinal Anclada puede simular otras afecciones neurológicas. Debe sospecharse este síndrome en un paciente que comience con deformidades ortopédicas de los miembros inferiores, de curso progresivo, independientemente de la edad de aparición.
Existe afectación en grado moderado de los estudios electrofisiológicos, los cuales contribuyen a detectar el grado de afectación neurológica y su topografía.
Definición.
Se define esta condición cuando el cono medular se encuentra anormalmente bajo, "Anclado” y fijo por un Filum Terminal Engrosado que genera invariablemente un déficit neurológico variable. Se asocia a otros casos más severos de Disrrafismo Espinal como el Lipomielomeningocele, el Meningocele, la Diastematomelia, los lipomas del Fillum Terminale, entre otros.
Embriología y patogénesis.
Hacia la 8° semana de gestación, la médula espinal embrionaria se extiende hasta la parte inferior del canal espinal. En esta fase de Embriogénesis comienza a aparecer el Filum Terminal que representa la involución de la médula espinal del 2° segmento coccígeo.
A partir de este momento, la médula espinal y las vértebras crecen longitudinalmente y la columna vertebral crece más rápido que la médula espinal. Esta progresión longitudinal , produce un ascenso de la médula espinal respecto al nivel vertebral. La proporción de ascenso de la médula espinal llega al máximo entre las 8 y 18 semanas.

Si el Cono Medular se fija a un Filum Terminal Inelástico entre las 8 y 25 semanas de gestación, el ascenso normal de la medula espinal se inhibirá, con la resultante posición baja del cono medular.
La distorsión anatómica de la médula espinal por la tracción se acompaña de una reducción significativa en el flujo sanguíneo medular asociado a disminución de la actividad Mitocondrial Neuronal.
Los procedimientos de liberación quirúrgica temprana de la Médula Anclada revierten estas perturbaciones metabólicas; por lo tanto el diagnóstico precoz evita las lesiones neurológicas irreversibles.
Manifestaciones clínicas.
Los siguientes son los aspectos clínicos más relevantes:
- Se presenta con igual frecuencia en hombres y mujeres.
- El 50% tienen una anomalía cutánea como un hemangioma, un hoyuelo o un Nevus Velloso en la región lumbar inferior.
- El inicio de los síntomas generalmente ocurre entre los 5 y 15 años. Son de tipo neurológico, ortopédico y urológico.
- Dentro de los síntomas predominantes se encuentran: debilidad motora progresiva y atrofia muscular, dolor lumbo-sacro que empeora con la flexión, pie cavo, escoliosis, trastornos de la marcha, poliuria, vaciamiento vesical incompleto y enuresis.
Estudios paraclínicos.
Tratamiento.
La liberación precoz de la Médula Anclada mediante sección quirúrgica del Filum Terminal es el tratamiento de elección
Siringomielia
La Siringomielia y la Médula Espinal Anclada Postraumáticas se pueden producir después de una
lesión en la médula espinal. Pueden aparecer desde dos meses hasta muchas décadas después de la lesión.
Los resultados puede ser devastadores, causando nuevos niveles de discapacidad mucho tiempo después de que una persona tuvo una rehabilitación satisfactoria. Los síntomas clínicos de la Siringomielia y la Médula Espinal Anclada son los mismos y pueden incluir el deterioro progresivo de la médula, la pérdida progresiva de sensibilidad o fuerza, sudor abundante, espasticidad, dolor y disreflexia autonómica (DA).
La Médula Anclada es una afección donde se forma tejido de cicatriz y ancla (o fija) la médula espinal a la dura, la membrana de tejido blando que la rodea. Este tejido cicatrizal impide el flujo normal del fluido espinal alrededor de la médula e impide el movimiento normal de la misma dentro de la membrana. El anclaje causa la formación de quistes. La Médula Anclada se puede producir sin que haya evidencia de Siringomielia, pero la formación quística postraumática no se produce sin algún grado de anclaje de médula.
Las imágenes por resonancia magnética (IRM) detectan fácilmente los quistes en la médula espinal, salvo que haya varillas, placas o fragmentos de proyectil.
Las Médulas Ancladas y la Siringomielia Postraumáticas se tratan quirúrgicamente. El ‘desanclaje’ implica una cirugía delicada para liberar el tejido cicatrizal alrededor de la médula para restaurar el flujo de líquido en la columna y el movimiento de la médula. Además, se coloca un pequeño injerto en el sitio del anclaje para fortificar el espacio dural y reducir el riesgo de que se vuelvan a formar cicatrices. Si hay un quiste, se coloca un tubo o desviación (shunt) dentro de la cavidad para drenar el fluido del quiste. La cirugía generalmente mejora la fuerza y reduce el dolor; no siempre devuelve la función sensoria perdida.
También puede haber parálisis de brazos y piernas. Los adultos y adolescentes con Malformación de Chiari que antes no mostraban ningún síntoma pueden mostrar signos de alteraciones progresivas, como movimientos oculares involuntarios y rápidos hacia abajo. Otros síntomas pueden incluir mareos, dolor de cabeza, visión doble, sordera y alteración en la capacidad de coordinar movimientos con episodios de dolor agudo dentro y alrededor de los ojos.